
WIEN2K 套裝軟體是目前使用密度泛函理論計算週期性體系的電子結構的最精確的計算程式之一。它在密度泛函理論框架下使用局域 (自旋)密度近似 (L(S)DA) 或廣義梯度近似 (GGA),採用全勢 (Full Potential, FP) 方法和 (線性)增廣平面波 ((L)APW)+ 局域軌道 (lo) 基組。WIEN2K套裝程式計算中對芯層電子考慮完全相對論效應,對價層電子考慮標量相對論效應。
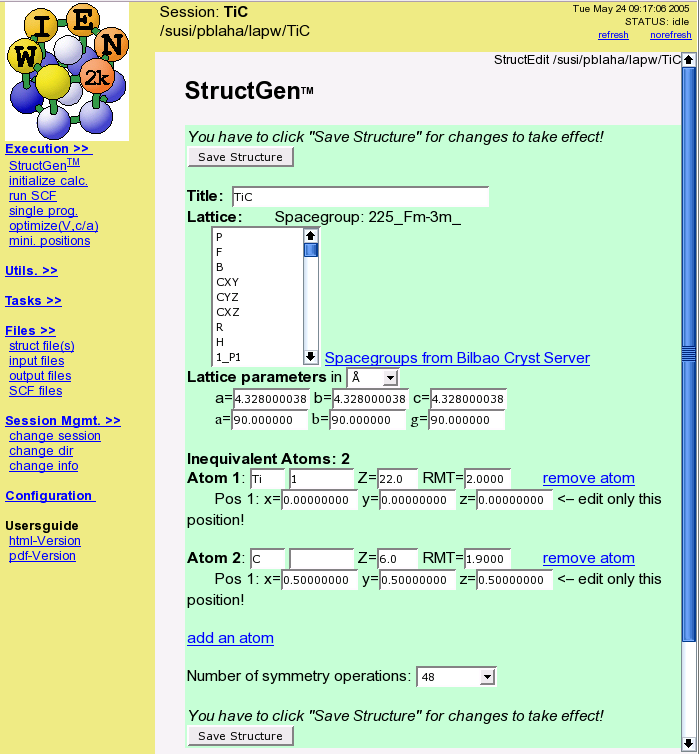
- Define your structure (cif-file import, spacegroup support, symmetry detection)
- initialize (semi-automatic guided input generation)
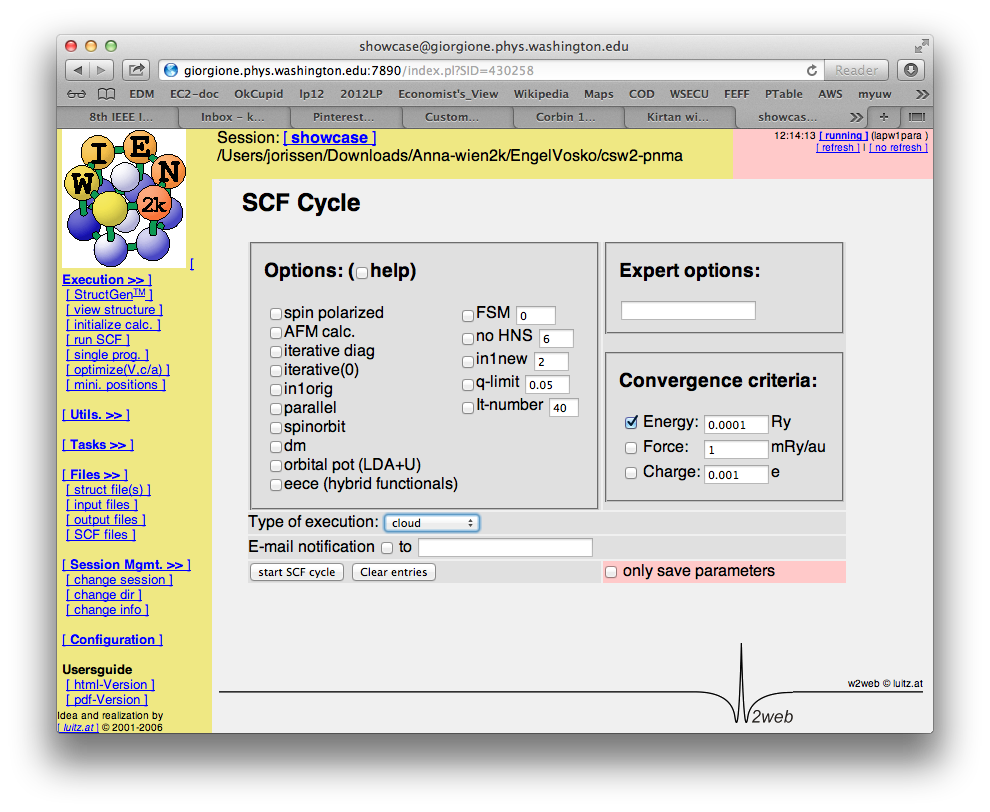
- run scf-cycle (with/without simultaneous optimization of atomic positions)
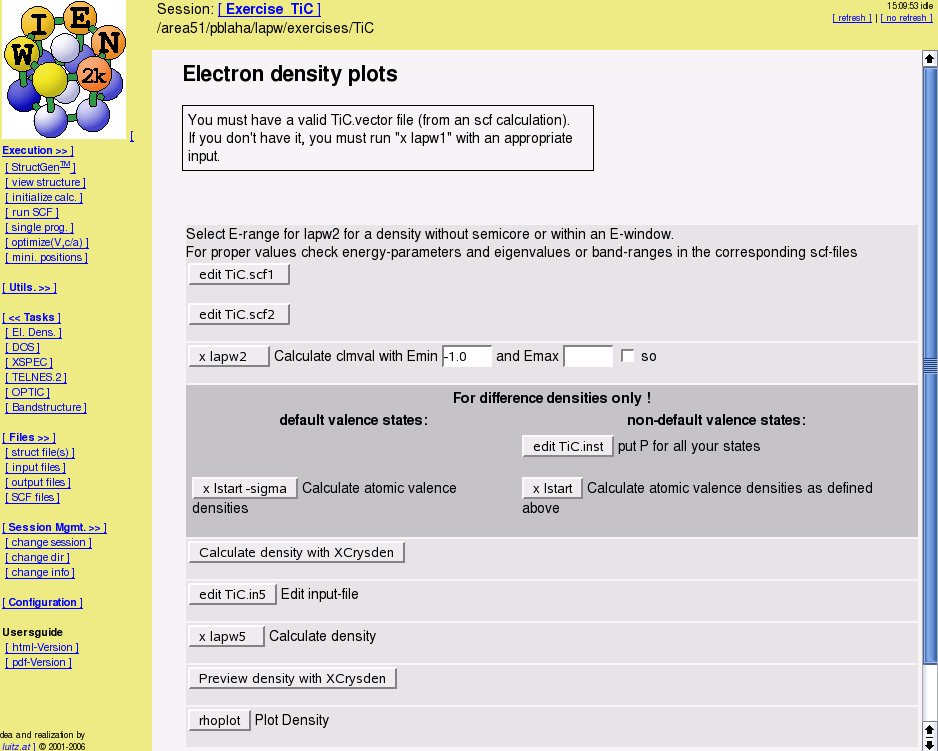
- Calculate some properties ("Guided Tasks" in w2web)
- write a publication (NOT yet supported in w2web, you must do it yourself)
更新介紹
Version 23.2 is a minor update but includes a VERY IMPORTANT bug-fix for lapw1. In WIEN2k_23.1 there is a severe bug in lapw1 for all "cubic" crystals (positive atom numbers in case.struct), which can affect severely the eigenvalues (and thus total energies and all other properties. Results for cubic systems in WIEN2k_23.1 are NOT reliable and update is required.
The program TRACK by Antik Sihi and Sudhir K. Pandey has been added to the "unsupported software page". It calculates TRAnsport properties for Correlated materials using the Kubo formalism. There are a few other new features/bug-fixes as compared to version 23.1 and update is highly recommended.
- SRC:
- hfpara_lapw: The program sumhfpara is not called for hybrid-NMR calculations.
- init_mgga_lapw: predefination of more mGGAs
- lapw1para_lapw: allows now hostnames with "." in .machines
- lapw2para_lapw: veccopy not done for hybrid-DFT calculations (since they are already local)
- qtlpara_lapw: missing label "single" defined
- restore_lapw: if restored in0 file is NOT a mGGA, rm case.vtau
- scf-cycles (run*_lapw): additional :TAUCTOxxx for taufile convergence printed to scf file
- x_lapw: x qtl -band (switch added in lapw2 -fermi step)
- x_nmr_lapw: support for mgga (vtau and vspmgga files copied)
- SRC_lapw0: inputpars.F, lapw0.F: small print modifications to scf0
- SRC_lapw1: important bug fix in atpar.F for cubic cases (see above)
- SRC_cif2struct: adding additional monoclinic spacegroups: param.inc,module.f,spacegroup.f,getsgnum.f,getlattype.f
- SRC_pairhess: patch.f: small fix for precise positions
- SRC_trig: Makefile updated to compile also write_inwplot
- 內置 230 個空間群圖形化使用者介面
- 方便的計算電子態密度和能帶結構
- 介面友好的網頁終端操作環境 (W2web),可方便地產生和修改輸入檔
- 自旋極化計算,方便精確計算鐵磁性和反鐵磁性結構
- 旋 - 軌耦合計算
- X 射線發射和吸收譜計算
- 簡單和複雜體系的結構優化,包括晶胞參數和原子內座標
- 固體的光學特性和電子能量損失譜計算
- 2011 版新增的 TP-MBJ 泛函可以在 DFT 理論下大大改進半導體材料的帶隙結構,與 GW 計算結果相近
- LDA, GGA, meta-GGA (libxc interface), LDA+U and EECE, orbital polarization, Hybrid-DFT
- centro- or non-centrosymmetric cells (mode), all 230 spacegroups built in
- spin-polarization (ferro- or antiferromagnetic structures), spin-orbit coupling
- sequential mode, k-parallel mode (without MPI, slow network with common NFS), massively parallel MPI mode (shared memory or Infiniband)
- Energy bands and density of states
- electron densities and spin densities, x-ray structure factors, potentials, STM and AFM simulations
- Baders's "atoms-in-molecule" concept
- total energy, forces, equilibrium geometries, structure optimization, elastic constants, molecular dynamics
- Phonons, with an interface to K.Parlinski's PHONON or A. Togos Phonopy program
- electric field gradients, isomer shifts, hyperfine fields, NMR chemical shifts, NMR Knight shifts
- x-ray emission and absorption spectra, electron energy loss spectra
- optical properties
- fermi surfaces
- The program is written in FORTRAN90 and runs under Linux/Unix on practically all platforms (single Linux-PCs to high-performance clusters, IBM RS6000, SGI ).
- The most efficient platform changes rapidly with time, although we expect that the best price/performance ratio will also in future be some Linux PC based on an Intel architecture with high memory bandwidth (Intel I7 architecture or Inter Xeons (more expensive); for current benchmark tests click here ). Install the Intel ifort compiler + Intels mkl-libarary (www.intel.com) or use gfortran + openblas.
- It requires at least 1 GB memory for small systems (to about 10 atoms per unit cell) or more for larger systems. At present we recommend a multicore CPU with 2-4 GB memory / core and twice as much swap space (don't forget to configure the latter!). We have treated systems up to 100 atoms per unit cell on workstations with large memory (16 GB) and with more than 1000 atoms/cell on clusters with 64 - 1024 cores and fast network. 1 GB (or 10-1000GB for large cases) of disk space is required.
- k-point parallelization is possible and highly efficient on a cluster of PCs (Gbit network is enough), provided a common NFS-file system is available and login (rsh or ssh) is configured properly.
- A fine grain parallelization for a single k-point is also available. It requires fast communication (shared memory or fast networks (Infiniband), Gb Ethernet is not really sufficient), MPI, FFTW and Scalapack.
- In order to use all options (including the graphical user interface or XCRYSDEN) the following public domain packages must be installed on your system: xcrysden, tcsh, ghostview (+png support), gnuplot (+png support), acroread (or similar), graphical www-browser (firefox), Perl, python (2.7 or higher), octave.
- Additional useful (optional) packages include: VESTA, Wannier90, libxc, phonopy.